导读
Wilson病是全球性遗传代谢性疾病,其发病率远比预估情况多见,致病基因携带率为1/(90-150),成年人发病率(有神经系统症状者)高达1/30 000。据报道,1993年11月至2015年12月,安徽中医学院神经病学研究所附属医院收治来自全国各地的Wilson病患者已超2万例。可见,我国Wilson病其实也并不少见。
作者:Peter Ferenci 维也纳医科大学
Wilson病(WD)又称肝豆状核变性,是由ATP7B基因突变引起的常染色体遗传性铜代谢障碍。铜是一种重要的微量元素,参与多种蛋白及金属酶的组成,也是许多器官,包括肝脏、骨骼、结缔组织、大脑和心脏的正常生长、发育和功能所必需的元素。但当铜的贮积超过细胞的安全储存水平时,就会导致细胞的损伤。
肝ATP7B蛋白的主要功能是将铜从细胞内转运至分泌途径,包括分泌至胆汁、或与无铜铜蓝蛋白结合形成功能性铜蓝蛋白。以此来调节铜在机体内的含量。ATP7B蛋白的缺乏或功能障碍则导致胆汁铜分泌下降,引起铜在肝在和肝外组织的沉积。以肝脏和大脑基底节受累最为严重。
WD若能及早确诊、及时服用排铜药物,即可减少损害。可同时,该病又常被忽略而延误诊断。若不及时接受治疗,特别是爆发性WD,死亡率高达95%。
Wilson病的特征
该病在任何年龄段均可发病,主要发生于3-35岁。患者临床表现复杂多样,主要特征为肝炎及肝硬化、神经精神异常、角膜K-F环及与急性肝衰竭相关的急性溶血。也可能无症状,而这些无症状患者通常由家系筛查发现。
典型的神经特征包括震颤、构音障碍(发音困难)、肌张力障碍;大约90%的患者有K-F环(见图1);约60%的患者有精神症状,且大多数患者存在肝组织学异常。不过,Wilson病患者的肝组织学改变不具特异性。
患者可在儿童期就表现为肝病症状,神经或精神症状会较晚使才表现出来。该病有明显的性别效应:肝病表现在女性中较常见,而神经表现则在男性中更为常见;约2/3的急性暴发性患者为女性。
目前已经发现了800多种不同的ATP7B突变,但WD的不同表型表现与特定突变无关。
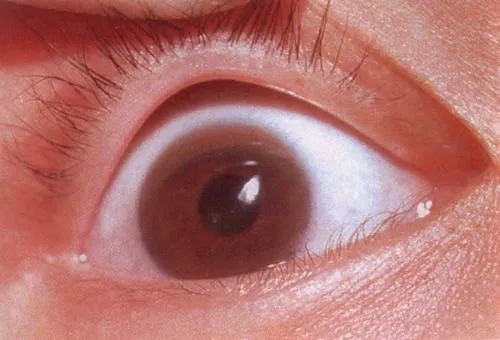
图1 1例Wilson病患者的K-F环(来源:《希夫肝脏病学》)
Wilson病的诊断
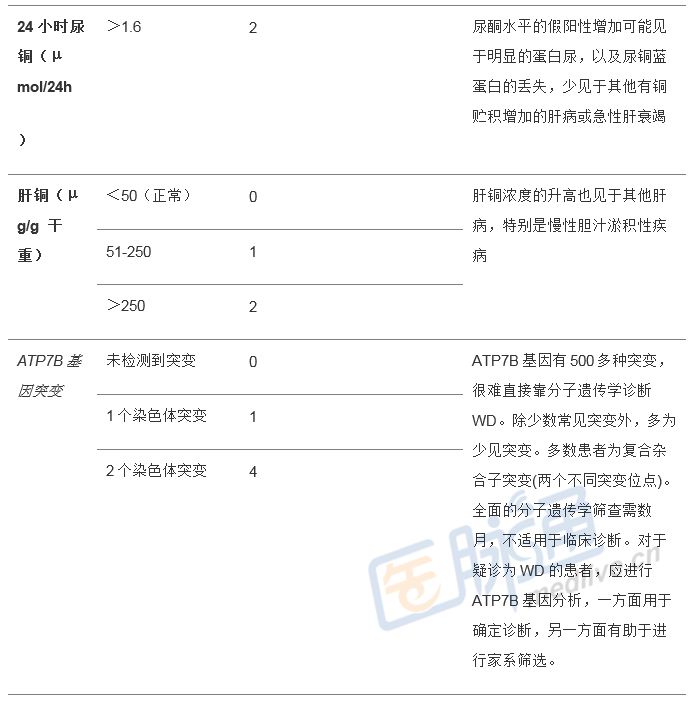
通常需要一系列测试,包括与铜代谢相关的测量(血清铜、铜蓝蛋白、尿铜排泄、肝铜含量),以及分子遗传学测试来诊断或排除WD。同时,血浆铜蓝蛋白通常会低于正常水平,「游离」铜含量升高。Leipzig评分(见表1)有助于WD的诊断。患者的直系亲属必须接受评估,要做到这一点,基因测试尤其具有价值。
表1 Leipzig评分(≥4分可确定诊断)

治疗
经确诊后,患者可终身使用铜螯合剂(D-青霉胺、曲恩汀、四硫代钼酸铵)或锌剂等,及时促进铜的排泄。在缺乏WD治疗药物疗效有力证据的情况下,建议使用铜螯合剂作为初始治疗手段。
治疗WD急性肝衰竭或失代偿期肝硬化的最有效方法是肝移植。而肝移植治疗神经性WD的效果有限,不推荐使用。
其他所有患者均可通过药物治疗达到正常预期寿命。
医脉通编译自:Peter Ferenci. Wilson’s disease: Fatal when overlooked, curable when diagnosed. J Hepatol. July 2019.
本文为医脉通编译整理,未经授权请勿转载至其他平台。欢迎读者分享至朋友圈。